












所有平台仅提供服务对接功能,所载文章、数据仅供参考,股市有风险,投资需谨慎,用户需独立做出投资决策,风险自担!

时间:2022-04-08 19:58:03来源:

艺术解释Gem目录中的微生物基因组序列如何有助于填补关于在地球微生物体中发挥关键作用的微生物的知识的差距。此图像补充了由世界各地环境样本产生的52,515个微生物草案的公共储存库,将已知的细菌和古代的普及性扩展为44%,现在可用,并描述于2020年11月9日在自然生物技术。从地球的微生物瘤目录的这种基因组目录中的工作来自涉及200多名科学家,DOE联合基因组研究所(JGI)和DOE系统生物学知识库(KBase)的研究人员的合作。
尽管在过去十年中排序技术和计算方法进展,但研究人员已经发现了基因组,只需一小部分地球的微生物普遍性。因为大多数微生物不能在实验室条件下培养,因此不能使用传统方法对其基因组进行测序。识别和表征行星的微生物普及是了解微生物作用在调节营养周期中的关键,以及对其在各种研究领域中可能拥有的潜在应用的见解。
从世界各地的环境样本产生的52,515个微生物草案的公共储存库,现在可在20世纪60年11月9日在自然生物技术中扩大细菌和古代的已知群体和古代的群体。被称为Gem(来自地球的微生物群体的基因组)目录,这项工作来自涉及200多名科学家,美国能源部的研究人员(DoE)联合基因组研究所(JGI)的研究人员,该科学用户设施位于劳伦斯伯克利国家实验室(伯克利实验室)和DOE系统生物学知识库(KBase)。
MetageNomics是使用各种处理,测序和分析的方法来研究环境样品中的微生物群落的研究,而不需要分离史式生物。“使用称为Metagenome Binning的技术,我们能够直接从测序的环境样品重建数千种组装的基因组(MAGS),而不需要在实验室中培养微生物,”斯蒂芬Nayfach,这项研究首次作者和研究科学家尼科斯·京核苷酸的微生物组数据科学集团。“这项研究真正脱颖而出的是我们分析的样本的显着环境普遍性。”
JGI Metagenome计划和高级作者的Emiley Eleo-Fadrosh阐述了Nayfach的评论。“这项研究旨在涵盖最广泛,最持持久的样品和环境,包括自然和农业土壤,人类和动物宿主相关的和海洋和其他水生环境 - 这非常出色。”
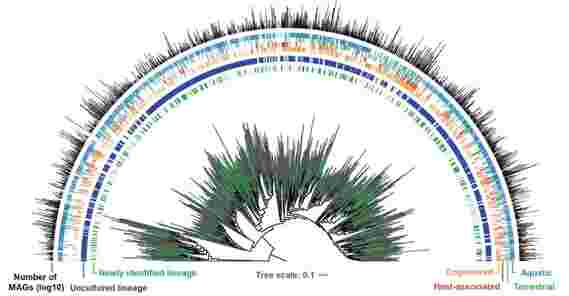
宝石目录扩展了在系统发育树上看到的细菌和古序命令,具有来自宝石目录(以绿色)和以前存在的参考基因组(以灰色为单位)的新裂缝的遗漏基因组谱系。在系统发育树周围,条带图表指示订单是否是未培养的(蓝色;仅由毕蛋白组合组装的基因组或MAG表示)或培养(灰色;由分离基因组表示)。接下来的四条条图表指示环境分布,而条形图表明从每个订单中恢复的宝石目录中的基因组数。
通过通过社区科学计划由JGI测序的环境样本中生成了大部分数据,并且已经在JGI的集成微生物基因组(IMG / M)平台上提供。Eloe-Fadrosh指出,“大数据”挖掘是一个很好的例子,以便通过公开提供数据来提高数据并增强价值。
要承认已经采样的调查人员的努力,根据JGI数据使用政策,Eleo-Fadrosh达到了世界各地的200多名研究人员。“我觉得承认从这些样本中收集和提取DNA的重大努力非常重要,其中许多来自独特,难以访问的环境,并邀请这些研究人员成为IMG数据联盟的一部分的共同作者,”她说。
利用这个大规模的数据集,Nayfach将MAG分类为18,000个候选物种组,其中70%是新颖的,而在此时可用的50万个现有基因组比较。“看着生活树,这是醒目的遗漏血统只由玛格所代表,”他说。“虽然这些草案是不完善的,但它们仍然可以揭示关于未培养的微生物的生物学和普遍性。”
研究人员的团队在利用基因组存储库中使用多次分析,并且IMG / M团队开发了几个更新和功能来挖掘宝石目录。(在Metagenome垃圾箱上观看这个IMG网络研讨会以了解更多信息。)一组用于次级代谢物生物合成基因簇(BGCS)的新型次生代谢物的数据集,将这些BGC在IMG / ABC(生物合成基因簇的地图集)中增加31%。(聆听这个JGI自然生殖集团的基因组矿业集。)Nayfach还与另一个团队合作,预测IMG / VR(病毒)和宝石目录的所有病毒之间的宿主病毒连接,将81,000病毒 - 70%尚未与主人相关联 - 23,000毫克。
根据这些资源,KBase,为生物学家和生物信息管理员设计的多机构协作知识创建和发现环境,为成千上万的MAG开发了代谢模型。该模型现在可以在公共叙述中提供,提供可共享的可重复的工作流程。“代谢建模是孤立基因组的常规分析,但尚未以未开垦的微生物的规模进行,”Eloe-Fadrosh“,”我们认为与KBase的合作会增加这些MAG的聚类和分析的价值。

从Artarctica的干谷收集的环境样本中IMG中的数据用于研究。
“只需将这个数据集带入KBase就可以立即价值,因为人们可以找到高质量的MAG并使用它们来告知未来的分析,”Argonne National实验室的KBase计算生物学家JoséP.Faria说。“构建代谢模型的过程很简单:您只需选择一个基因组或MAG,然后按一个按钮从生物化反应和注释之间的映射数据库中构建模型。我们查看在基因组中的注释和所产生的模型,以评估生物体的代谢能力。“(在代谢建模上观看这个KBase网络研讨会。)
KBase用户订婚领先Levela Wood-Charlson补充说,通过从Gem数据集中展示了代谢模型的轻松,Metagenomics研究人员可能会考虑分支进入这个空间。“大多数偏心组合研究人员可能不愿意才能培养一个完全新的研究领域[代谢建模],但它们可能对生物化学如何影响他们的工作原因感兴趣。基因组学社区现在可以使用KBase的简单路径从基因组或Mags来探讨新陈代谢,以便建模,“她说。
格鲁吉亚理工学院Kostas Konstantinidis,其中一个共同作者之一,其数据是目录的一部分,“我不认为有许多机构可以做这种大规模的偏心神经,并且具有大规模的能力分析。这项研究的美丽是它在这种规模完成的是,史化实验室不能做,并且它使我们对微生物普遍性和功能的新见解。“

来自藻类样品的IMG中的数据用于研究。
他已经找到了在他自己的研究中利用目录的方法,了解微生物如何应对气候变化。“通过这个数据集,我可以看到找到每个微生物的位置以及它有多丰富。这对我的工作和其他人进行了类似的研究非常有用。“此外,他有兴趣扩大参考数据库的普及,他正在开发称为微生物基因组图集,以允许通过添加MAGS来进行更强大的分析。
“这是社区的一个很好的资源,”康斯坦辛迪斯补充道。“这是一个数据集,随后将有助于更多的研究。我希望JGI和其他机构继续做这种项目。“
参考:“地球微生物的基因组目录”由Stephen Nayfach,Simon Roux,Rekha Seshadri,Daniel Udwary,Neha Varghese,Frederik Schulz,东营吴,David Paez-Espino,I-Min Chen,Marcel Huntiagan,Krishna Palaniappan,Joshua Ladau,Supratim Mukherjee,TBK Reddy,Torben Nielsen,Edward Kirton,JoséP.Faria,Jokaka N. Edirisinghe,Christopher S. Henry,Sean P. Jungbluth,Dylan Chivian,Paramvir dehal,Elisha M. Wood-Charlson,Adam P. Arkin,Susannah G. Stone,Axel visel,IMG / M数据联盟,Tanja Woyke,Nigel J. Mouncey,Natalia N. Ivanova,Nikos C. kyrpides和Emiley A. Eloe-Fadrosh,2020年11月9日,自然Biotechnology.doi:
10.1038 / s41587-020-0718-6
该工作还使用了国家能源研究科学计算中心(NERSC)的资源,位于伯克利实验室的另一个DOE科学用户设施办公室。
声明:文章仅代表原作者观点,不代表本站立场;如有侵权、违规,可直接反馈本站,我们将会作修改或删除处理。





![面向生物技术的基于细菌的纳米工厂[视频]](http://www.jjchem.net/uploadfile/2021/0616/2021555531.jpg)
图文推荐

2022-04-08 19:58:03

2022-04-08 18:58:03

2022-04-08 17:58:03

2022-04-08 16:58:03

2022-04-08 15:58:03
2022-04-08 14:58:03
热点排行
精彩文章

2022-04-08 18:58:05
2022-04-08 17:58:06

2022-04-08 16:58:06
2022-04-08 15:58:04
2022-04-08 14:58:07

2022-04-08 11:58:04
热门推荐
