





所有平台仅提供服务对接功能,所载文章、数据仅供参考,股市有风险,投资需谨慎,用户需独立做出投资决策,风险自担!

时间:2022-09-19 14:06:51来源:网络整理
温馨提示:本推文共25篇,预计阅读时间25分钟左右。请选择您感兴趣的。文末会有过去三周周报的链接。
1. 自然。能源:分散在碳纳米管上的镍酞菁分子旨在实现高选择性的二氧化碳减排
CO2 的电化学还原是可持续燃料生产的一条有前景的途径。一个巨大的挑战是开发低成本、高效的电催化剂以实现高转化率和高产物选择性。在本文中,一系列负载在碳纳米管上的酞菁镍分子被设计为分子分散的电催化剂(MDE)。具有甲氧基官能团的优化 MDE 解决了原始镍酞菁催化剂的稳定性问题,并在 -300 mA cm-2 的高电流密度的气体扩散电极装置中实现大于 99.5% 的选择性。 CO2 到 CO。它在 -150 mA cm-2 下稳定运行 40 小时。 MDEs 的明确活性位点也有助于从原位 X 射线吸收光谱以及影响电催化性能的结构因素的理论计算中深入了解机理。
原文链接:
2. 自然。 Commun.:高价金属位点可激活晶格氧,增强OER活性
阳极析氧反应(OER)被认为是水电解的动力学瓶颈。具有高价态的过渡金属位点可以加速反应动力学,从而提供固有的高活性,但面临热力学形成的障碍。本文展示了通过原子间电子相互作用在多组分 FeCoCrNi 合金薄膜表面重建的(羟基)氢氧化物中高度氧化的 Ni 物种的精细设计。光谱和理论研究表明,Fe成分能够形成Ni4+物种,并且Ni2+→Ni3+→Ni4+的多步演化在能量上是有利的。动态构建的 Ni 物种将空穴驱动到氧配体中,以促进分子内氧偶联并触发晶格氧活化,形成 Fe-Ni 双位点,成为具有高本征活性的最终催化中心。在碱性电解液中 300 mV 的过电位下,表面重组的 FeCoCrNi 催化的 OER 反应表现出 3601 A gmetal-1 的出色质量活性和 0.483 s-1 的 TOF。
原文链接:
3.焦耳:Cu-Ag 串联催化剂用于 CO2 高速电解制多碳产品
串联电催化通过多组分催化剂设计将化学复杂途径中的各个步骤解耦,这一概念对于将 CO2 电还原为多碳物质 (C2+) 具有吸引力,尤其是在高速率下。本文表明,气体扩散电极 (GDE) 上的 Cu-Ag 串联催化剂可以通过在 Ag 上将 CO2 还原为 CO 以及随后在 Cu 上的碳耦合来提高 CO2 生成 C2+ 的速率。在 1 M KOH 中,在 -0.70 V vs. RHE 的电位下,添加 Ag 后,Cu 表面 C2+ 部分的电流密度从 37 mA/cm2 增加到 160 mA/cm2,并且两种金属的区别是没有相互干扰的。此外,在纯 CO2 或 CO 气氛下,串联催化剂的固有 C2H4 和 C2H5OH 活性显着高于单独使用 Cu 的活性。 Ag 创造的富含 CO 的局部环境可以促进 C2+ 在 Cu 上的形成,而不仅仅是提供 CO2 或 CO,为串联催化剂在三相环境中的反应提供新的见解。
原文链接:
4. J. 上午。化学。 Soc.: 单原子催化剂中的金属比反应性:在 MgO 上原子分散的 4d 和 5d 过渡金属上的 CO 氧化
了解和调整原子分散金属在氧化物上的催化性能是合理开发单原子催化剂 (SAC) 的主要途径,而结构规则的 SAC 系列的设计和合成也提供了作为金属功能的系统研究properties 提供广泛的想法。在通用 MgO 载体上合成了一系列基于各种 4d(Ru、Rh、Pd)和 5d(Ir、Pt)过渡金属的单原子催化剂。补充实验(X 射线吸收光谱)和理论研究(密度泛函理论)表明膦酰基,两种金属阳离子都优先占据台阶边缘下方的八面体配位 MgO 晶格位置,因此受到氧化物载体的高度限制。在暴露于氧气耗尽的 CO 氧化条件后,FTIR 光谱显示在预催化温度下 CO 驱动的单原子金属中心部分划界,然后在稳态条件下形成表面碳酸盐物质。这些发现得到了 DFT 计算的支持,表明表面金属突起的驱动力和最终结构是金属相关的,但在所有情况下,八面体配位的 M4+ 碳酸盐作为等效状态。实验确定的表观反应活化能在 96 ± 19 kJ/mol 的范围内,其中 Pt 导致最低能垒。结果表明,对于 SACs 中的单原子位点,通过金属选择可实现的 CO 氧化反应性差异小于先前通过氧化物载体上相邻氧化还原中心的机械接合所证明的差异,这表明氧化物表面的化学调控与支持金属的选择。
原文链接:
5. ACS Catal.:原子分散的 Ru 催化剂,通过缔合机制将氮低温活化为氨
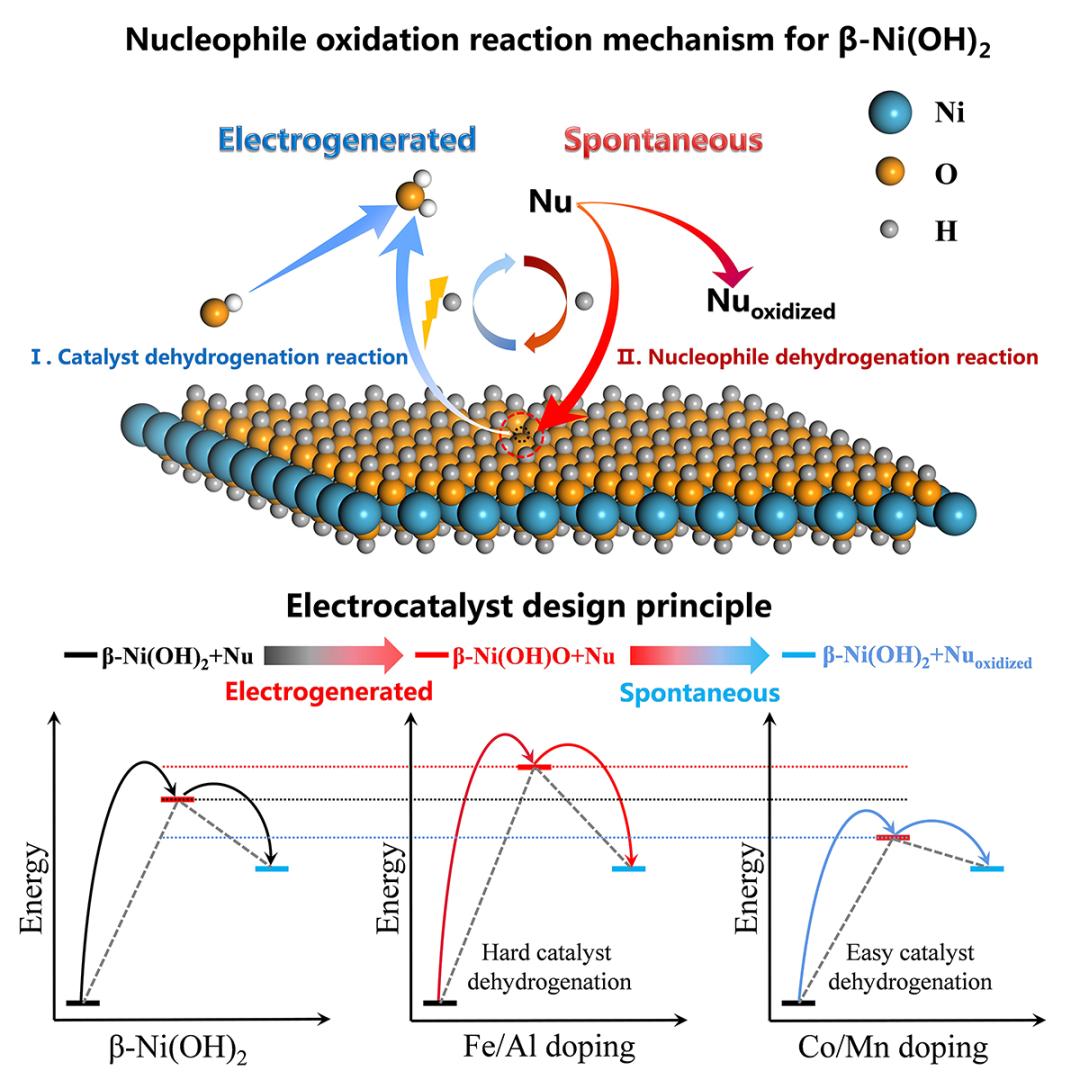
使用铁基或钌基催化剂合成 NH3 通常需要苛刻的反应条件。均质的单原子Ru中心,其中Ru原子分别锚定在HZ的微孔中,在温和条件下通过缔合途径有效促进NH3的合成。基于同步加速器的近边缘 X 射线吸收精细结构 (NEXAFS) 和原位 DRIFTS 分析表明 =N- 基团是主要中间体,DFT 计算进一步表明,与钌纳米团簇不同,单个钌原子具有氢原子在 HZ 中导致 N 氢化而不是直接 N 解离 , 间接 N-N 键解离更有可能通过形成 NHNH3* 中间体发生。 N-N 键断裂的能垒从 *N2 的 2.90 eV 一直下降到 *NHNH3 >04 eV 的 0.@,这表明 N2 加氢是显着削弱N-N 键。此外,决定速率的步骤从 N≡N 三键的解离转变为 *N2H2 的形成。最终,0.2 wt %Ru/H-ZSM-5 催化剂的 NH3 合成率是迄今为止报道的 Ru 基催化剂最高的 (1.26 mol NH3gRu–1 h-1)@ > 在 300°C 和 1 MPa)。
原文链接:
6.科学:来自大自然的灵感,将地球上丰富的元素作为高效催化剂
地球上富含金属的金属酶催化了生命所必需的许多氧化还原转化。相比之下,铂族金属由于其高活性、热稳定性和对化学毒物的耐受性,几十年来已广泛用于许多工业催化反应。本文推测了自然界中各种反应的基本原理,这些反应可以极大地扩大地球丰富的金属在催化中的用途。这突出了这些金属区别于贵金属的关键物理特性。然后看看自然界,了解如何利用它们的内在特性来生产高效催化剂,这些催化剂对于燃料和化学品的可持续生产和转化至关重要。
原文链接:
7. Mater.Today:增强光催化水分解的二维光催化剂和可调支持
可持续制氢越来越受到关注,可见光驱动的水分解被认为是最有前景的制氢和太阳能储存方法之一。近几十年来,在温和条件下筛选了不同的材料,二维(2D)层状材料被认为是光催化水分解反应的良好候选者。二维单层MoS2在各种催化体系中显示出其潜力,近年来也被用于光催化水分解反应。
然而,目前的研究表明,MoS2单层的内在活性较低,阻碍了其实际应用。这是由于光激发载流子在室温下快速复合,导致量子效率(QE)差。在此,我们报告了一种通过将 2D MoS2 纳米片与固态极化面载体相结合来延长激子寿命的最先进策略。极化小面载流子引入的强局部极化可以促进电荷分离过程,并通过改变具有不同表面极性的载流子进行调整。极性氧化物表面暴露于末端氧高能晶面,已知这些晶面会产生垂直于表面的净偶极矩。载体的这种不同表面性质将导致上述金属的不同金属-载体相互作用。由此产生的复合结构显示出可见光驱动的光催化水分解反应的极大增强,以在高温下从纯水以化学计量比 2:1 生成氢和氧。延长的激子寿命提高了光催化性能,并且通过掺钌的 MoS2 纳米片获得了 2977 μmol g-1h-1 的超高析氢活性,在极性二氧化铈载体上具有令人印象深刻的 QE,这是迄今为止报道的最佳结果之一对于同类的催化系统来说还很遥远。更令人兴奋的是,还观察到激子寿命与局部极化之间的线性关系,这表明合理的光催化剂设计可以通过引入极化面材料来简单地设计其局部极化。
原文链接:
8. 高级。功能。材料:具有可调带结构和电荷转移特性的类静脉g-C3N4的合成用于选择性光催化H2O2生产
与传统的蒽醌法或电催化法相比,双电子氧还原光催化制H2O2作为一种环保策略受到广泛关注。在此,我们报告了一种仿生叶脉状 g-C3N4 作为 H2O2 生产的有效光催化剂,具有可调带结构、优化的电荷转移和选择性双电子 O2 还原。结合实验和理论计算研究了能带结构和电荷转移的调控机制。 CN4 (287 mol h-1)@> 的 H2O2 产率大约是原始 CN (87 mol h-1)@>) 的 3.3 倍,而 H2O2 的表观量子产率由CN4 在 420 nm) 产率达到 27.8%,远高于目前许多其他光催化剂。这项工作不仅为设计具有优异 H2O2 生产效率的光催化剂提供了新的策略,而且有助于理解缺陷和掺杂 深入了解位点在光催化活性中的作用。
原文链接:
9. 自然。 Commun.: 石墨化磷配位 Fe 单原子加氢转化
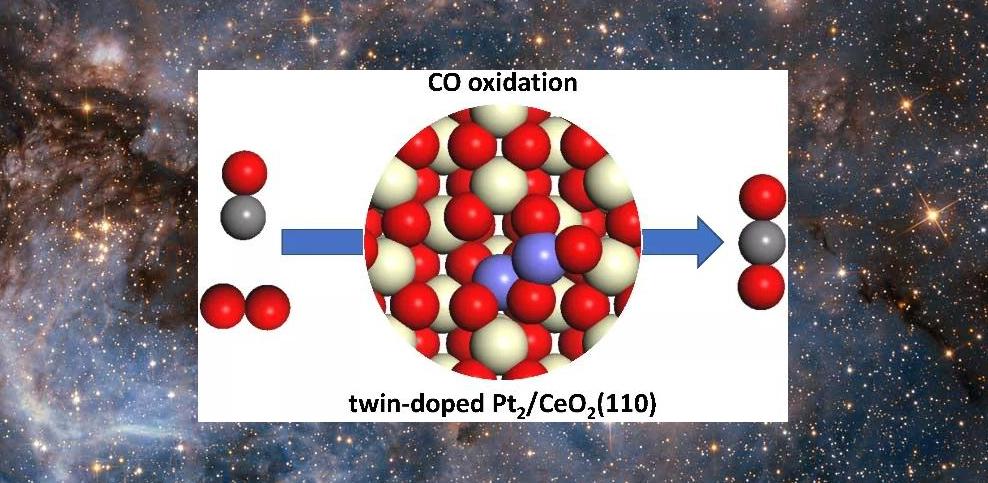
单原子金属-氮-碳(M-N-C)催化剂引起了广泛的兴趣,然而,原子分散的金属-磷-碳(M-P-C)催化剂的开发尚未实现,尽管分子金属-膦配合物已经已广泛应用于均相催化。在这里,我们成功地在P掺杂碳上构建了石墨磷配位的单原子Fe,在N-杂环的多相氢化、硝基芳烃的官能化、还原胺化反应性能和反应普遍性方面表现出优异的催化性能,而相应的原子分散Fe在相同的反应条件下,嵌入 N 掺杂碳上的原子几乎没有活性。此外,我们还发现,当浸渍的铁原子转化为铁簇/纳米粒子时,石墨磷配位单原子铁的催化活性显着降低。该工作不仅对设计以P原子为配位点的单原子催化剂具有重要意义,也对M-P-C催化剂在多相催化中的应用具有重要意义。
原文链接:
10. J. 上午。化学。 Soc.: 高特异性类氧化酶MOF-818纳米酶催化儿茶酚氧化
尽管纳米酶已被广泛研究,显示其性能优于天然酶和传统人工酶,但高度特异性纳米酶的开发仍然是一个挑战。儿茶酚氧化酶专门催化儿茶酚氧化成相应的邻苯醌,对黑色素和其他多酚类天然产物的生物合成很重要。在本研究中,我们首先提出MOF-818含有一个模拟天然儿茶酚氧化酶活性位点的三核铜中心,具有高效的儿茶酚氧化酶活性、良好的特异性和无类过氧化物酶的特点。 MOF-818作为一种新型的儿茶酚氧化酶纳米酶,具有良好的特异性和较高的催化活性。
原文链接:
11. 安吉。化学。诠释。编:含有稳定自由基的 MOF 催化的太阳能固氮:结构转变引起的效率提高
在此,我们提出了一种新型的紫精基金属有机骨架 (RMOF) 含 Gd-IHEP-7,它在空气中加热后会发生单晶到单晶的生长。转化为生成 Gd-IHEP-8。由于良好的自由基-自由基相互作用,两种 RMOF 都表现出良好的空气和水稳定性,并且它们的长寿命自由基在 200-2500 nm 范围内产生宽光谱吸收。 Gd-IHEP-7 和 Gd-IHEP-8 在太阳能驱动的固氮方面表现出良好的活性,氨生成率分别为 128 和 220 mmol h-1 g-1。实验和理论计算表明,两种 RMOF 具有相似的固氮途径。 Gd-IHEP-8相对于Gd-IHEP-7的催化效率提高归因于中间体通过增强的氢键稳定。
原文链接:
12. J. 上午。化学。 Soc.: 通过安装高效光催化连接器将活性位点精确嵌入介孔 Zr 框架
在过去的二十年里,微孔金属有机框架 (MOF) 的孔隙工程得到了广泛的研究,各种官能团已扩展到各种框架中。然而,可靠地获得具有目标孔径和首选功能的 MOF 并不常见。这一点尤其重要,因为许多精心设计的材料的适用性通常受到微孔框架的小孔径的限制。在这里,我们设计并合成了一种基于 Zr6 簇和四羧酸配体的介孔 MOF,命名为 PCN-808。 PCN-808 具有易于合成的不饱和金属结构,框架固有的灵活性使 PCN-808 成为通过接头进行修饰后合成修饰的主要支架。线性钌基金属配体已成功且精确地安装在 PCN-808 的开孔壁中,同时保持框架的中孔率。对所得材料PCN-808-BDBR在氮杂-亨利反应中的光催化活性进行了考察,6次催化循环后转化率较高。此外,由于骨架的介孔性质,PCN-808-BDBR在光催化氧化双氢青蒿素制备青蒿素方面也表现出优异的收率。
原文链接:
13.安吉。化学。诠释。主编:光催化分散可逆失活自由基聚合制备直链和支链含氟聚合物
拓扑会影响聚合物的特性和应用。因此,已经做出了相当大的努力来控制拓扑。在这项工作中,作者开发了一种基于可逆失活自由基聚合(RDRP)的光催化发散合成方法,该方法能够同时制备支化度可调和链端保真度高的低-直链和支链含氟聚合物。该方法有利于通过扩链光-RDRP生成复杂结构(如项链状和拖把状含氟聚合物),为获得具有高锂离子转移数和良好离子电导率的含氟聚合物电解质提供了一种新的通用平台膦酰基,这将为先进材料工程创造更好的机会。
原文链接:
14. J. 上午。化学。 Soc.: NHC 稳定的锗离子:催化 CO2 功能化反应活性和效用
第一个没有受体酰基离子的重锗类似物,[RGe(O)(NHC)2]X (R = MesTer = 2,6-(2,4,6-Me3C6H2) 2C6H3; NHC = IMe4 = 1,3,4,5-四甲基咪唑-2-亚基;X = (Cl or BArF = {(3,5–(CF3)2C6H5)4B}), 分离得到通过 [RGe(NHC)2]X 与 N2O 反应,锗酰亚胺离子被转化为第一个单给体稳定的锗酯 [(NHC)RGe(O)(OSiPh3)],以及相应的重类似物([RGe(S)(NHC)2]X 和 [RGe(Se)(NHC)2]X),表现出典型的类酰基行为。利用极性末端 GeO 在 CO2 的锗酰离子键活化中和硅烷, 前者被发现是 CO2 可逆活化的一个例子, 并模仿了过渡金属氧化物的行为. 使用 CO2 作为 C1 源的胺的 CO2 氢化硅烷化和胺 N-官能化的还原 它是化学反应中的活性催化剂, 这证明了它的过渡金属特性。从实验和计算两个方面进行的机理研究 r表明该反应是由 NHC-siloxygermylene [(NHC)RGe(OSiHPh2)] 介导的。
原文链接:
15. 安吉。化学。诠释。主编:氮掺杂碳辅助乙烯氧氯化制氯乙烯的一锅串联反应
0.0@>
在这里,作者展示了 CuCl2/Al2O3 和 N 掺杂碳的双功能催化剂的设计理念,用于在 250°C 和环境压力下以高达 76 的高产率进行高效的一锅乙烯氧氯化工艺生产 VCM %,高于传统工业两步法(约50%)。除了第一方面使用的CuCl基乙烯氧氯化催化剂外,作者还证明了第二方面的无金属氮掺杂碳上含有N-官能团的活性位点同时催化乙烯氧氯化和二氯乙烷。 EDC) 脱氯化氢。得益于 N 掺杂碳的双重功能,通过 EDC 脱氯化氢和乙烯氧氯化作用的表面 Cl* 循环加剧了 VCM 的形成。这两种反应都通过氧氯化作用原位消耗表面 Cl* 来增强,其中 Cl* 由 EDC 脱氯化氢产生。这项工作为在温和条件下在单程反应器中通过乙烯氧氯化生产 VCM 的可行替代方案铺平了道路。
0.1@>
原文链接:
16. 安吉。化学。诠释。 Ed.: 用于化学选择性加氢的高密度和热稳定性钯单原子催化剂
0.3@>
单原子催化剂 (SAC) 在许多与能源和环境相关的反应中表现出出色的活性和/或选择性,但它们在高位点密度和还原气氛下的稳定性仍未得到解决。本文探讨了还原气氛下高位点密度钯单原子(高达 5 wt%)的内在驱动力及其对加氢反应的独特催化性能。原位实验(包括扫描透射电子显微镜、近环境压力 X 射线光电子能谱、X 射线衍射和准原位同步加速器 X 射线吸附)和理论计算表明,在渗碳过程中,Pd 原子倾向于氧化晶体转变为碳化物晶体并迁移到表面富含空位的碳化钼表面,导致钯原子的表面富集而不是颗粒形成。 Pd1/α-MoC催化剂在取代硝基芳烃液相加氢(>99%)和CO2气相加氢制CO(>98%)中表现出高活性和良好选择性,远优于其纳米类似物。更重要的是,Pd1/α-MoC 催化剂可以承受高达 400 oC 的苛刻还原/反应气氛,而没有任何可观察到的单原子聚集。这项工作为寻找单原子催化剂提供了一条新途径,即使在苛刻的还原条件下也能在高位点密度下实现更好的性能和稳定性。
0.4@>
原文链接:
17. 自然。材料:翅片分子筛催化剂
0.6@>
越来越多的证据表明,合成纳米沸石具有显着降低内部扩散约束以增强催化和吸附性能的优势。然而,生产尺寸小于 100 nm 的沸石晶体并非易事,通常需要使用复杂的有机物,并且通常会导致产品收率降低。在这里,作者提出了一种替代方法,通过在种子上外延生长鳍状突起来增强沸石的传质性能。作者证明了这种方法在两种常见沸石上的可行性,并证实翅片与下面的晶种进行晶体配准,并且二次生长不会阻碍进入微孔。翅片沸石的分子建模和时间分辨滴定实验探测了内部扩散,揭示了质量传递的显着改善,这与模型反应的催化实验结果一致,表明这些结构表现为准纳米晶体,尺寸类似于那些鳍。大小可比。该方法可以推广到其他沸石和硅铝酸盐材料的合理合成。
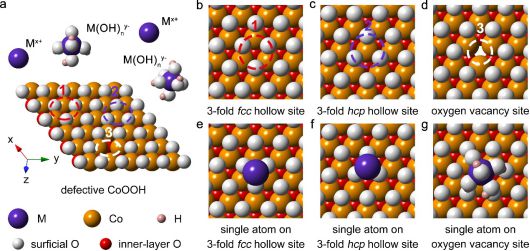
0.7@>
原文链接:
18.化学:CO2辅助选择性裂解乙烷中C-C/C-H键的界面活性位点
0.9@>
将页岩气中未充分利用的乙烷选择性升级为温室气体 CO2 可通过干重整(C-C 键断裂)生产合成气或通过氧化脱氢(C-H 键断裂)生产乙烯。然而,由于负载催化剂的复杂性,识别乙烷中选择性键断裂的活性位点仍然具有挑战性。在本文中,通过结合动力学测量、原位表征和计算,研究了 CeO2 负载催化剂上的乙烷-CO2 反应,以揭示不同界面位点的功能。 Pd/CeOx界面负责提供活性氧,缺电子氧Pd表面的分子物种促进非选择性键断裂生成合成气,FeOx/Pd界面上的富电子氧增强选择性断裂C-H 键生成乙烯。目前的工作确定了使用不同界面结构升级页岩气和二氧化碳的机会。
原文链接:
19. 自然。通讯:二氧化铈原子铜(I/II)位点对分子氧的吸附与活化
支持的原子金属位点具有离散的分子轨道。精确控制这些位点的能量是获得具有优异选择性的新型反应途径的关键。在这里,作者利用铈 (Ce) 阳离子框架来降低孤立铜 (Cu) 位点的 3d 轨道能量,以实现选择性氧 (O2) 活化。操作 X 射线吸收光谱、电子顺磁共振和密度泛函理论模拟表明,[Cu(I)O2]3-选择性吸附分子 O2,在 298 K 时形成很少报道的亲电 η2-O2。在辅助最终还原过程中,η2-O2 与相邻的 Ce(III) 阳离子结合到两个O2-,在453 K时形成两个Cu-O-Ce氧桥。与CuO簇相比,孤立的Cu(I)/(II)位点对CO的氧化作用更好,活性高出十倍。在 373 K 和 0.01 bar PCO 时,跃迁频率为 0.028±0.003 s-1。[Cu(I)O2]3- 独特的电子结构表明其潜力选择性氧化。
原文链接:
20. 物质:具有优异催化效率的分级孔沸石单晶
作为尺寸和形状选择性催化剂,沸石广泛用于石油和精细化工过程。然而,它们的微孔严重阻碍分子扩散并且对焦炭的形成很敏感。具有完全互连、有序和可调的多尺度孔隙率的分级孔隙沸石单晶是理想的解决方案。然而,它们的合成仍然极具挑战性。在这里,作者报告了一种通用的受限沸石结晶过程来实现这些优异的性能。在大分子反应和甲醇-烯烃反应中,这种沸石单晶通过缩短扩散长度而提供显着改进,同时保持形状选择性特性,使它们提高催化活性和寿命,大大减少焦炭形成,并降低失活率传质。它们的工业应用可以导致设计出高效和最低能耗的创新反应器和工艺。
原文链接:
21. 安吉。化学。诠释。主编:在 Pd-P 阴极上以 H2O(D2O) 为 H(D) 源的炔烃选择性转移半氢化反应
使用廉价且安全的氢源开发一种简便且可控的路线以选择性地将炔烃半氢化为具有特定构型的烯烃是一项巨大的挑战。本文报道了在 Pd-P 合金阴极上使用 H2O (D2O) 作为氢源在较低电位下对各种末端和内部炔烃进行选择性半氢化(氘化)。密度泛函理论 (DFT) 计算表明,P 掺杂增强了炔烃的比吸附,并增强了水电解产生吸附原子氢 (H*ads) 的内在活性。炔烃的半氢化可以在较低的电位下完成,烯烃产物的选择性高达 99%,法拉第效率高达 78%,优于纯钯和商业钯/碳。炔烃的这种电化学半氢化可以通过 H*ads 加成途径而不是质子耦合电子转移过程进行。在较低电位下,Pd-P 对 Hadd 的还原和对 C≡Cπ 键的优先吸附比对 C≡C 部分的吸附更有利,导致烯烃选择性好,容易获得热力学不稳定但合成显着的 Z- Z 选择性大于 99% 的烯烃。该方法能够合成更有用但更具挑战性的单氘、二氘和三氘烯烃,氘掺入率高达 99%。在Pd-P|NiSe双电极体系中,双电极法生产关键工业原料己二腈和阴极烯烃显示出良好的应用前景。
原文链接:
22. 高级。能量。材料:一锅法合成CO-吡啶衍生催化剂,在Nafion基膜电极组件中将CO2高效电化学转化为CO
为了实现二氧化碳的实际利用,迫切需要开发一种具有高法拉第效率(FE)、能量效率(EE)和电流密度的电化学二氧化碳还原反应(CO2RR)工艺。本文报道了一种简便的一锅法合成钴基和聚 4-乙烯基吡啶基催化剂,用于在基于 nafion 的膜电极组件的析氢反应中将 CO2RR 转化为 CO,该催化剂可用于 pH 值的工作范围为 2 到 7。电池优化导致 CO2 转化为 CO,在 85 mA cm-2 下具有 92% FE 和 58% EE,这些特性归因于促进吡啶部分几乎均匀的合成和加工条件Co在纳米级配位,产生催化所需的适当配合物。优异的性能与简便方法的可扩展性和组件的可访问性相结合,为电化学 CO2RR 的商业化铺平了道路。
原文链接:
23. 高级。功能。材料:碳化钼-碳化钨双金属与氮掺杂碳的杂化:合理设计多孔复合纳米线以提高析氢催化活性
在本文中,开发了一种环保且稳定的方法来构建由 N 掺杂碳与双金属碳化钼 (MoxW2−xC) 杂化组成的自支撑单片电极,以形成用于析氢反应性 (HER) 的纳米结构) 复合纳米线。 MoxW2-xC与N掺杂碳的杂化可以有效调节复合纳米线的电催化性能,赋予N掺杂和MoxW2-xC掺入产生的大量活性位点,N掺杂碳基体具有优异的导电性, 同时,利用热力学有利的氢吸附自由能 (ΔGH*) 来正确定位 d 带中心以实现高效的析氢催化,从而形成无粘合剂的三维自支撑整体电极。该电极具有可接近的纳米孔、理想的化学成分和稳定的复合结构。通过调整 Mo/W 比,碳布上的最佳纳米线 Mo1.33W0.67C@NC 实现了低过电位(酸性和碱性环境中电流密度分别为 115 和 108 mV)为 10 mA cm−2),具有较小的 Tafel 斜率,分别为 58.5 和 55.4 mV dec-1,可保持稳定的性能 40 小时,性能超过大部分的HER电催化剂是基于金属碳化物的。
原文链接:
24. 安吉。化学。诠释。主编:钯催化的烯丙醇/CO直接选择性合成己二酸和其他二元酸
本文报道了一种通过钯催化烯丙醇二羰基化直接合成二酸的通用方法,包括工业上重要的己二酸。具体而言,PdCl2与双膦配体(HeMaRaphos)的结合可以促进两种不同的羰基化反应,具有高活性和良好的选择性。
原文链接:
25. 自然。催化剂:钯/手性降冰片烯协同催化构建轴向手性化合物
Axial chiral biaryls are common structural motifs in functional materials, biologically active natural products, pharmaceuticals, and chiral catalysts/ligands. Therefore, the efficient preparation of these special scaffolds is an important task in the field of organic chemistry. Here, we report a general modular platform technique for the construction of axial chirality via palladium/chiral norbornene synergistic catalysis. It is a three-component cascade reaction process with readily available aryl iodides, 2,6-substituted aryl bromides and alkenes (or alkynes, boronic acids, etc.) as reactants. A variety of substrates with various functional groups (88 examples) are compatible with this method. Other features include a unique stereo-induced model, excellent enantioselectivity, step economy and scalability. This method is also applicable to the synthesis of chiral fluorenols via axial-to-center chirality transfer at high stereochemical fidelity. This work is expected to have broad applications in the synthesis of chiral ligands and the design of asymmetric catalytic catalysts.
原文链接:
声明:文章仅代表原作者观点,不代表本站立场;如有侵权、违规,可直接反馈本站,我们将会作修改或删除处理。






图文推荐

2022-09-19 14:05:09

2022-09-19 13:49:06

2022-09-19 12:36:30

2022-09-19 10:57:14

2022-09-19 10:14:14

2022-09-19 10:04:23
热点排行
精彩文章
2022-09-19 14:06:51

2022-09-19 13:49:09

2022-09-19 13:06:14

2022-09-19 13:06:05

2022-09-19 10:14:06

2022-09-19 09:31:17
热门推荐
